The field of cellular biology is currently undergoing a revolution in data generation. The goal of my research in proteomics is to develop statistical models which help answer basic biological questions on protein function and expression through the use of genome scale datasets.
Protein Translation & Codon Usage Bias

Comparison between observed and expected protein production rates for 5847 verified genes in the S. cerevisiae genome.
Codon usage bias, i.e. the non-uniform usage of synonymous codons within a gene sequence, is a ubiquitous biological phenomenon. Although these subtle patterns held within a gene may not seem very intriguing, they actually allow us to
develop and apply fundamental concepts in evolutionary biology, such as Wright’s Fitness Landscapes, while simultaneously extract information on gene expression from coding sequences themselves. My work in this area began a number of years ago with a simple model of ribosome movement along an mRNA [9]. This model allowed us to explore the role of nonsense errors (errors which lead to premature termination of protein translation) as a selective force on the evolution of codon usage bias. On a more fundamental level, this model also allowed us to develop a simple framework for linking genotype, codon usage, to phenotype, protein production costs. We can use this framework to predict the protein production rate of a given gene [13].

Distribution of NAI values for S. cerevisiae S288c and simulated genomes. Solid line represents null expectation.
These predictions are essentially based on the degree of adaptation a gene displays to minimize the cost of protein production through its codon usage. We have used these same concepts to develop the only biologically based measure of codon usage bias, the Nonsense Adaptation Index (NAI) [17]. This work has provided us a foundation for understanding the nature of different selective forces, such as nonsense errors and the cost of ribosome usage. In addition, we’ve begun expanding our modeling framework to include tRNA competition effects and intra-ribosomal tRNA-mRNA stability. One important result for this work is that missense errors are unlikely to be the primary selective force driving the evolution of codon bias, overthrowing one of the most favored hypothesis for codon bias [21].
Protein Complex Composition
One of the central goals of proteomics is to determine the function of each protein. Because most proteins function in the context of a protein complex, one of the first steps towards identifying protein function is to correctly identify protein complex composition. Working with Andreas Wagner and Laura Salter, I have developed a Bayesian framework for inferring protein complex composition from high-throughput protein interaction datasets [5]. We have applied this framework to two high-throughput datasets which use similar affinity purification techniques for identifying yeast protein complexes.
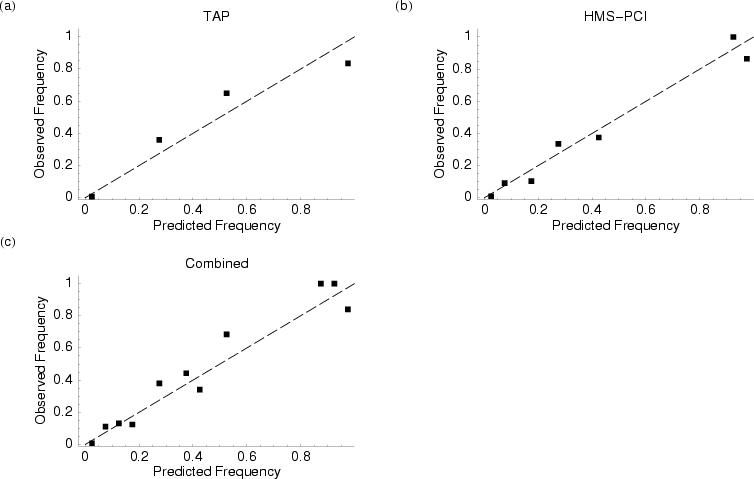
Predicted vs. observed frequencies of protein-protein associations based on the (a) TAP, (b) HMS-PCI, and (c) the combined datasets. Dashed line illustrates an ideal 1: 1 correlation between observed and expected frequencies.
My framework is based on a probabilistic model of how the data is actually generated. The approach I have developed has the distinct advantage that it can assess the quality of a dataset based on its internal self-consistency. Our results indicate that affinity purification based techniques miss 50 to 80 % of all proteins in a complex and usually include two or three additional, non-complex proteins. These high error rates makes it hard to have much confidence inferring complex composition from a single experiment. However, because our approach is Bayesian in nature we can incorporate information on protein complexes from multiple experiments, including those from different datasets. The result is that we are able to calculate the probability two proteins are in the same complex to a surprising degree of accuracy.
